

Aktuell: EU-Kommission will bei der MDR nachbessern
Es ist soweit! Ab nun gelten die Anforderungen der EU-Verordnung 2017/745 (MDR).
Um die Verfügbarkeit von Medizinprodukten auf dem EU-Markt aufrechtzuerhalten, plant die EU-Kommission Nachbesserungen an der EU-Verordnung 2017/745 (MDR).
Unser MDR Ticker hält Sie auf dem laufenden. Oder kontaktieren Sie uns per Email. Wir schicken Ihnen gerne neuen Informationen, sobald sie uns vorliegen.
Durchführung und Erstellung der Risikomanagementakte im Rahmen der MDR (2017/745)
Risikomanagement und MDR
Regulatorisches zum Risikomanagement
Um sicherzustellen, dass serienmäßig hergestellte Produkte den Anforderungen der Medical Device Regulation 2017/745 (MDR) jederzeit entsprechen und dass die Erfahrungen, die im Zuge der Verwendung der hergestellten Produkte gesammelt werden, in das Herstellungsverfahren einfließen, sollten alle Hersteller laut MDR, Vorwort, Abschnitt 32 über ein Qualitätsmanagementsystem und ein System zur Überwachung nach dem Inverkehrbringen verfügen, das der Risikoklasse und der Art des betreffenden Produkts angepasst sein sollte. Zur Minimierung des Risikos bzw. um Vorkommnisse im Zusammenhang mit Produkten zu verhindern, sollten die Hersteller des Weiteren ein Risikomanagementsystem und ein System für die Meldung von Vorkommnissen und Sicherheitskorrekturmaßnahmen im Feld einrichten. Das Risikomanagement sollte ferner sorgfältig mit der klinischen Bewertung des Produkts abgestimmt und darin berücksichtigt werden, was auch für die klinischen Risiken gilt, denen im Rahmen der klinischen Prüfungen, der klinischen Bewertung und der klinischen Nachbeobachtung nach dem Inverkehrbringen nachzugehen ist. Das Risikomanagement und die Verfahren der klinischen Bewertung sollten miteinander verknüpft sein und regelmäßig aktualisiert werden (MDR, Vorwort Abschnitt 33).
In Anhang I, Kapitel 1 der MDR werden die allgemeinen Anforderungen hinsichtlich der Risiken eines Medizinproduktes gelistet. Unter anderem heißt es dort, dass die Hersteller ein Risikomanagementsystem einführen, dies umsetzen, es dokumentieren und fortschreiben. Das Risikomanagement ist dabei als kontinuierlicher iterativer Prozess während des gesamten Lebenszyklus eines Produkts zu verstehen, der eine regelmäßige systematische Aktualisierung erfordert. Bei der Durchführung des Risikomanagements müssen die Hersteller u. a. (MDR, Anhang I, Kapitel 1, Absatz 3):
- einen Risikomanagement-Plan für jedes Produkt festlegen und dokumentieren,
- die bekannten und vorhersehbaren Gefährdungen, die mit jedem Produkt verbunden sind, identifizieren und analysieren,
- die Risiken, einschätzen und bewerten, die mit der bestimmungsgemäßen Verwendung verbunden sind und die bei einer vernünftigerweise vorhersehbaren Fehlanwendung auftreten,
- die Auswirkungen der in der Fertigungsphase und, insbesondere durch das System zur Überwachung nach dem Inverkehrbringen gewonnenen Informationen, auf Gefährdungen und deren Häufigkeit, auf Abschätzung der verbundenen Risiken sowie auf das Gesamtrisiko, das Nutzen-Risiko-Verhältnis und die Risikoakzeptanz bewerten, und
- erforderlichenfalls auf der Grundlage der Bewertung der Auswirkungen die Kontrollmaßnahmen gemäß den Anforderungen anpassen.
Für die Einrichtung des Risikomanagements sollten sich die Hersteller hierfür an die harmonisierten Normen halten. Die MDR, Vorwort Abschnitt besagt hierzu, dass Angesichts der wichtigen Rolle, die der Normung im Bereich der Medizinprodukte zukommt, sollten die Hersteller die Konformität mit den in dieser Verordnung festgelegten grundlegenden Sicherheits-, Leistungs- und sonstigen rechtlichen Anforderungen, beispielsweise an Qualitäts- und Risikomanagement, durch Einhaltung der harmonisierten Normen gemäß der Verordnung (EU) Nr. 1025/2012 des Europäischen Parlaments und des Rates (15) nachweisen können.
Für das Risikomanagement wären das die EN ISO 14971, auch wenn diese Norm noch nicht als harmonisierte Norm für die MDR geführt wird. Die EU hat jedoch eine Liste mit Normen veröffentlicht, die die Normen enthält, die sie harmonisieren will. Hierunter fällt auch die EN ISO 14971. (Link Liste: https://ec.europa.eu/docsroom/documents/43584?locale=en)
Risikomanagement-Prozess
Die EN ISO 14971 besagt, dass ein Hersteller einen Prozess festlegen, implementieren, dokumentieren und aufrechterhalten muss, um so:
- die mit dem Produkt verbundenen Gefährdungen und Gefährdungssituationen zu identifizieren;
- die damit verbundenen Risiken einzuschätzen und zu bewerten;
- diese Risiken zu beherrschen;
- die Wirksamkeit der Maßnahmen zur Risikobeherrschung zu überwachen.
Der Risikomanagementprozess kann Teil eines Qualitätsmanagementsystems sein, z. B. eines, das auf der ISO 13485:2016 basiert. Dies wird von EN ISO 14971 aber nicht gefordert.
Der Risikomanagement-Prozess muss eine Risikoanalyse, eine Risikobewertung, die Risikobeherrschung und Aktivitäten / Daten und Informationen während der Herstellung und den nachgelagerten Phasen der Produktion enthalten.
Neben der EN ISO 14971, kann der Technische Report ISO/TR 24971 als Leitfaden herangezogen werden.
Risikomanagement Plan (RM-Plan)
Der Risikomanagementplan beschreibt den Umfang der Risikomanagementaktivitäten, die Verantwortlichkeiten und Befugnisse der Beteiligten, die Akzeptanzkriterien von Risiken, die für das Medizinprodukt zu sammelnden und zu überprüfenden Informationen über die Produktion und die Zeit nach der Produktion sowie alle Risikomanagementaktivitäten, die während des gesamten Produktlebenszyklus durchgeführt werden. Der RM-Plan wird während des gesamten Lebenszyklus des Produkts überprüft und bei neu verfügbaren Informationen aktualisiert. Dabei sollte der Umfang der Aktivitäten dem mit dem Medizinprodukt verbundenen Risiko angemessen sein.
Risikoanalyse
Der Prozess der Risikoanalyse besteht aus der Beschreibung der bestimmungsgemäßen Verwendung des Medizinprodukts und der vernünftigerweise vorhersehbaren Fehlanwendung, der Ermittlung der sicherheitsrelevanten Merkmale des Medizinprodukts, der Identifizierung der mit dem Medizinprodukt verbundenen Gefahren und gefährlichen Situationen und der Abschätzung der Risiken für jede gefährliche Situation.
Laut ISO/TR 24971 ist eine Gefahr eine potenzielle Quelle für einen Schaden. Je nach der spezifischen Situation können Gefahren unterschiedliche Ursachen wie z. B. Elektrizität, bewegliche Teile, infektiöse Bakterien und Viren, Chemikalien, scharfe Kanten, usw. haben. Medizinprodukte verursachen nur dann Schäden, wenn eine Folge von Ereignissen eintritt, die zu einer gefährlichen Situation führt, die dann einen Schaden verursacht oder zu einem Schaden führt. Die Abfolge von Ereignissen kann eine chronologische Reihe von Ursachen und Wirkungen sowie Kombinationen von gleichzeitigen Ereignissen sein. Gefährliche Situationen können aber auch dann auftreten, wenn keine Fehler vorliegen, d. h. im Normalzustand des Medizinproduktes, wenn es wie vorgesehen funktioniert.
Für jede dieser identifizierten Gefährdungssituationen muss der Hersteller das zugehörige Risiko / die Risiken einschätzen. Die Risikoeinschätzung schließt eine Analyse der Wahrscheinlichkeit des Auftretens eines Schadens und des Schweregrades des Schadens ein (EN ISO 14971). Die Risikoeinschätzung kann qualitativ oder quantitativ erfolgen. Hierfür können mehrere Methoden, wie z. B. die PHA, FMEA und FTA angewendet werden.
Die Risikoanalyse setzt sich also aus der Identifikation möglicher Gefährdungen durch das Medizinprodukt und der Abschätzung von Wahrscheinlichkeiten, Schweregrad und damit der Risiken. Um die Risiken einschätzen zu können, sollte der Hersteller vorher festlegen, welche Risiken als akzeptabel und welche als inakzeptabel erachtet werden. Dies erfolgt zumeist in Form einer Risikobewertungsmatrix (Risikoakzeptanzmatrix). Beispiele für Risikobewertungsmatrizen oder Diagramme finden sich z. B. in der ISO/TR24971.
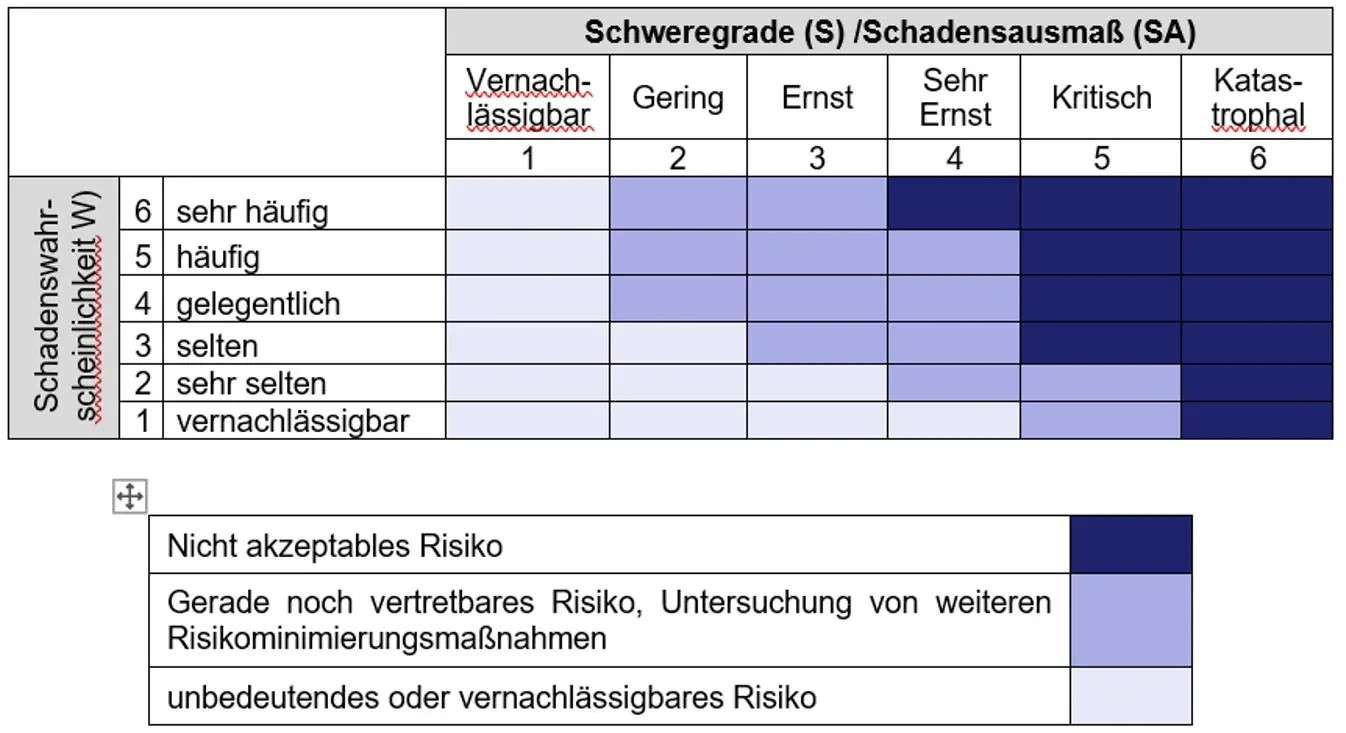
Für jede Art von Medizinprodukt (oder Medizinproduktfamilie) können spezifische Kriterien festgelegt werden, abhängig von ihren Merkmalen und ihrer Zweckbestimmung, oder es können für alle Medizinprodukte die gleichen Kriterien gelten. Die Kriterien für die Akzeptanz von Risiken werden im Risikomanagementplan festgehalten.
Risikobewertung
Bei der Risikobewertung vergleicht der Hersteller die eingeschätzten Risiken mit den vorher im Risikomanagementplan festgelegten Akzeptanzkriterien für die Zulässigkeit von Risiken und stellt fest, ob diese Kriterien erfüllt sind oder nicht.
Risikobeherrschung
Sollte während der Risikoanalyse festgestellt werden, dass es inakzeptabel Risiken gibt, muss er diese Risiken minimieren. Generell gilt, dass der Nutzen des Medizinproduktes, die Risiken überwiegen muss, die Risiken müssen also beherrscht werden.
Bei der Risikominimierung muss der Hersteller laut ISO EN 14971 dabei nach der folgenden Reihenfolge vorgehen:
- inhärent sicheres Design und sichere Herstellung;
- Schutzmaßnahmen im Medizinprodukt selbst oder im Herstellungsprozess;
- Informationen für die Sicherheit und, soweit zutreffend, die Schulung von Anwendern.
Dabei gilt, dass bei Maßnahmen zur Risikobeherrschung der Schweregrad des Schadens und / oder die Schadenswahrscheinlichkeit gemindert werden können. Erklärung zu den einzelnen oben genannten Punkten sind in der ISO/TR 24971 enthalten. Die Maßnahmen zur Risikominimierung müssen in der Risikomanagement- Akte dokumentiert werden.
Wenn der Hersteller während der Analyse der Optionen zur Risikobeherrschung bestimmt, dass die Minderung des Risikos nicht realisierbar ist, muss der Hersteller eine Nutzen-Risiko-Analyse für das Restrisiko durchführen (EN ISO 14971). Im Anschluss müssen die Maßnahmen ergriffen werden und die Wirksamkeit der getroffenen Maßnahmen muss überprüft werden. Die Ergebnisse dieser Verifizierung müssen wiederum in der RM-Akte dokumentiert werden.
Restrisken
Nach der Durchführung der Risikominimierungsmaßnahmen muss der Hersteller die Restrisiken erneut überprüfen. Unter Restrisiken versteht man laut EN ISO 14971 die Risiken, die nach der Umsetzung der Risikominimierungsmaßnahmen verbleiben. Hierfür werden wieder die im Risikomanagement-Plan aufgestellten Akzeptanzkriterien herangezogen. Das Restrisiko ist erneut entweder akzeptabel oder inakzeptabel. Wenn es als nicht akzeptabel eingestuft wird, sollten weitere Optionen zur Risikokontrolle untersucht werden. Ist eine weitere Risikokontrolle nicht durchführbar, kann eine Nutzen-Risiko-Analyse durchgeführt werden.
Beachten muss man ebenfalls, dass durch die getroffenen Maßnahmen zur Risikokontrolle weitere Risiken entstehen können, die dann ebenfalls wieder anhand der aufgestellten Akzeptanzkriterien bewertet werden müssen und eventuell weitere Risikominimierungsmaßnahmen nach sich ziehen.
Nutzen-Risikoanalyse
Die ISO 14971:2019 erlaubt es dem Hersteller, eine Nutzen-Risiko-Analyse für diejenigen Risiken durchzuführen, die anhand der im Risikomanagementplan festgelegten Kriterien als nicht akzeptabel eingestuft werden und für die eine weitere Risikobeherrschung nicht praktikabel ist. Wenn ein Restrisiko unter Anwendung der im Risikomanagementplan festgelegten Kriterien als nicht akzeptabel beurteilt wird und weitere Maßnahmen der Risikobeherrschung nicht durchführbar sind, darf der Hersteller Daten und Literatur (z. B. Daten zu Vergleichsprodukten auf dem Markt, dem Stand der Technik, usw.) zusammenstellen und bewerten, um zu bestimmen, ob der Nutzen der Zweckbestimmung dieses Restrisiko überwiegt.
Bewertung des Gesamt-Restrisikos
ISO EN 14971 verlangt, dass das Gesamtrestrisiko im Verhältnis zum Nutzen der bestimmungsgemäßen Verwendung des Medizinprodukts bewertet wird. Hierfür müssen sowohl die Akzeptanzkriterien des Gesamtrestrisikos, als auch die Methode zur Bewertung des Gesamtrestrisikos in den Risikomanagementplan aufgenommen werden.
Die Bewertung des Gesamtrestrisikos ist der Punkt, an dem das Restrisiko aus einer umfassenden Perspektive betrachtet wird. Alle identifizierten Gefahrensituationen wurden bewertet und alle Risiken wurden auf ein akzeptables Niveau reduziert oder auf der Grundlage einer Nutzen-Risiko-Analyse akzeptiert. Nun prüft der Hersteller, ob das mit dem Medizinprodukt insgesamt verbundene Restrisiko die Kriterien für die Annehmbarkeit des Gesamtrestrisikos erfüllt. Die Bewertung kann zu dem Schluss führen, dass das Medizinprodukt sicher ist. Wenn das Gesamt-Restrisiko im Verhältnis zum Nutzen zu hoch ist, darf der Hersteller weitere Risikominimierungsmaßnahmen ergreifen oder Änderungen am Medizinprodukt oder seiner Zweckbestimmung vornehmen.
Risikomanagement-Bericht (RM-Bericht)
Die ISO EN 14971 verlangt, dass die Endergebnisse des Risikomanagementprozesses überprüft werden, um sicherzustellen, dass der Risikomanagementplan ordnungsgemäß ausgeführt wurde, dass das Gesamtrestrisiko akzeptabel ist und dass geeignete Methoden zur Sammlung und Überprüfung relevanter Produktions- und Nachproduktionsinformationen vorhanden sind. Die Überprüfung des Risikomanagements erfolgt nach der Umsetzung und Überprüfung aller Risikokontrollmaßnahmen, jedoch vor der kommerziellen Freigabe des Medizinprodukts. Die Ergebnisse dieser Überprüfung werden im Risikomanagement-Bericht aufgeführt. Falls in z. B. Produktionsphase oder den nachgelagerten Phasen neue Informationen zur Verfügung stehen, die die Sicherheit des Medizinprodukts betreffen, kann es erforderlich sein, den RM-Bericht zu aktualisieren.
Hierbei unterstützen wir Sie gerne, kontaktieren Sie uns!
Quellen:
Risikomanagement von Medizinprodukten
Regulatorisches zum Risikomanagement
Unter Risikomanagement versteht man einen Prozess, um so die mit Ihrem Medizinprodukt verbundenen Gefährdungen und Gefährdungssituationen zu identifizieren; die damit verbundenen Risiken einzuschätzen und zu bewerten, diese Risiken zu beherrschen und die Wirksamkeit der Maßnahmen zur Risikobeherrschung zu überwachen. Hierbei können wir Sie folgendermaßen unterstützen:
- Erstellen /Überprüfen / Aktualisieren des Risikomanagement-Plans, Aufstellen der Akzepttanzkriterien
- Datenbank-Recherchen: Vorkommnissen (FDA MAUDE und TPLC, BfArM, EUDAMED (zukünftig), usw.)
- Durchführen / Aktualisieren / Überprüfen / Unterstützen bei der Risikoanalyse
- Erstellen und Aktualisieren des Risikomanagement-Berichts
Mit unserer Hilfe können Sie:
- Unnötige Arbeiten vermeiden
- Ihre Risikoanalyse regelmäßig an neu auftretende Risiken anpassen
- Zeit und Kosten sparen, indem Sie die aufwendige Arbeit an einer Stelle bündeln und bewerten lassen!
- Wir fertigen Ihre Dokumente schnell und zuverlässig in Deutsch oder Englisch an und nutzen dazu entweder Ihre oder die AnCura-eigenen normenkonformen Formblättern
- Gemeinsam finden wir Lösungen, Ihre Zufriedenheit ist unser oberstes Ziel!
Generell bieten wir unsere Leistungen sowohl in Deutsch, als auch in Englisch an.
Haben wir Ihr Interesse geweckt, dann kontaktieren Sie uns. Rufen Sie uns an, schreiben Sie uns oder füllen Sie unser Kontaktformular aus. Wir freuen uns auf Sie.