

Klinische Bewertung
Durchführung von klinischen Bewertungen nach der MDR (2017/745) und MedDev 2.7/1 Rev.4
Klinische Bewertung von Medizinprodukten - Regularien
Die klinische Bewertung
Mit Hilfe der klinischen Bewertung müssen alle Hersteller von Medizinprodukten (Klasse I bis III) nachweisen, dass Nutzen, Leistung und Sicherheit der Produkte in ausreichendem Maße gegeben sind. Es muss also sichergestellt werden, dass das Produkt weder den klinischen Zustand, noch die Sicherheit der Patienten oder von Dritten beeinträchtigt. Jedes Medizinprodukt muss daher entweder noch die grundlegenden Anforderungen der MDD oder aber die grundlegenden Sicherheits- und Leistungsanforderungen der MDR erfüllen.
Dokumentation einer klinischen Bewertung
Eine klinische Bewertung setzt sich aus den folgenden Dokumenten zusammen:
- dem klinischen Bewertungsplan (CEP, clinical evaluation plan) inkl. dem klinischen Entwicklungsplan (falls nötig) und
- dem klinischen Bewertungsbericht (CER, clinical evaluation report).
Wie die klinische Bewertung aufgebaut sein sollte und welche Inhalte sie enthalten muss, ist in zahlreichen Vorschriften und Normen beschrieben. Hierunter fallen u.a. der Anhang X der Medizinprodukterichtlinie (MDD); Artikel 10 und 61, sowie der Anhang XIV der EU-Verordnung 2017/745 (MDR), der Paragraph 19 des Medizinproduktegesetzes (MPG) und die MEDDEV 2.7.1.
Neben dem CEP und dem CER muss die Dokumentation noch eine Reihe weiterer Dokumente enthalten. Art und Umfang dieser Dokumentation richtet sich nach der Klasse des Medizinproduktes (siehe Abbildung). Die Risikomanagementakte sollte hier z. B. ebenfalls dazugezählt werden, da das Risikomanagement eine wesentliche Voraussetzung für die klinische Bewertung ist und umgekehrt.

Klinische Daten
Klinische Daten bilden die Grundlage einer jeden klinischen Bewertung. Anhand dieser Daten wird bewertet, ob das Medizinprodukt sicher und leistungsfähig ist und der Hersteller kann zudem beurteilen, ob die auftretenden Risiken im Zusammenhang mit der Anwendung in einem angemessenen Verhältnis zum erwarteten Nutzen des Produktes stehen. Die Daten können aus zahlreichen Quellen stammen, wie z. B.:
- Ergebnisse klinischer Prüfungen (durch den Hersteller)
- Wissenschaftlich relevante Fachliteratur über das Produkt oder die ausgewählten Äquivalenzprodukte (z.B. Ergebnisse klinischer Studien oder Artikel über sonstige klinische Erfahrungen mit dem Produkt)
- Klinische relevante Ergebnisse aus der Überwachung nach dem Inverkehrbringen (Post Market Surveillance (PMS) und Post Market Clinical Follow Up (PMCF))
- Meldungen über Vorkommnisse mit dem Produkt oder den Äquivalenzprodukten in den entsprechenden Datenbanken (BfArM, FDA (MAUDE und TPLC)).
Benannte Stellen erwarten von den Herstellern, dass diese mehrere Quellen für die klinische Bewertung heranziehen. Der Umfang des klinischen Nachweises muss sich aber an den Merkmalen des Produktes und an dessen Zweckbestimmung orientieren. Der Umfang für ein Klasse-I-Standard-Produkt beispielsweise aus dem Infektionsschutz kann geringer sein, als für ein Klasse-III-Produkt. Von den Herstellern wird daher verlangt, dass sie den Umfang der ausgewählten klinischen Daten (klinischer Nachweis) spezifizieren und begründen. Sie müssen zudem belegen, dass die ausgewählten klinischen Daten ausreichend sind, um die grundlegenden Sicherheits- und Leistungsanforderungen zu erfüllen. (Quelle: https://www.bundes¬gesundheits-ministerium.de/naki.html).
Kann man auf eine klinische Bewertung verzichten?
Weder die Medizinprodukterichtlinie (MDD), noch die EU-Verordnung 2017/745 (MDR) gestatten den Verzicht auf eine klinische Bewertung. Unter bestimmten Voraussetzungen kann man jedoch auf die klinische Bewertung anhand von klinischen Daten verzichten. Dies trifft in der Regel aber nur auf absolut unkritische Produkte zu.
Die MDR besagt dazu in Artikel 61, Absatz 10:
Wird der Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen auf der Grundlage klinischer Daten für ungeeignet erachtet, ist jede solche Ausnahme auf der Grundlage des Risikomanagements des Herstellers und unter Berücksichtigung der besonderen Merkmale des Zusammenspiels zwischen dem Produkt und dem menschlichen Körper, der bezweckten klinischen Leistung und der Angaben des Herstellers angemessen zu begründen; dies gilt unbeschadet des Absatzes 4. In diesem Fall muss der Hersteller in der technischen Dokumentation gemäß Anhang II gebührend begründen, warum er den Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen allein auf der Grundlage der Ergebnisse nichtklinischer Testmethoden, einschließlich Leistungsbewertung, technischer Prüfung („bench testing“) und vorklinischer Bewertung, für geeignet hält.
Dies trifft in der Regel aber nur auf absolut unkritische Produkte und sogenannte Legacy Devices (Altgeräte) und / oder well-established technologies zu. Laut MDCG 2020-6 kann die Konformität dieser Produkte mit den generellen Sicherheits- und Leistungsanforderungen der MDR kann dann unter anderem anhand der Äquivalenz mit anderen Produkten, Bewertung des Stands der Technik, einschließlich der Bewertung klinischer Daten von ähnlichen Produkten (similar devices), Beschwerden und Vigilanzdaten, pro-aktiv gesammelten PMS-Daten und Fallstudien zum Produkt nachgewiesen werden. Für Legacy devices der Klasse III und implantierbare Produkte, welche nicht unter die die well-established Technologies gehören, sollte die Konformität anhand von Daten aus klinischen Prüfungen und wissenschaftlichen Studien erfolgen.
Unter Legacy device (Altprodukte) versteht man laut MDCG 2020-6 alle Produkte, die zuvor gemäß der europäischen Richtlinie 93/42/EWG über Medizinprodukte (MDD) oder der Richtlinie 90/385/EWG über aktive implantierbare medizinische Geräte (AIMDD) gekennzeichnet wurden. Der Begriff well-established technology wird in der MDR erwähnt, aber nicht definiert. Laut MDCG 2020-6 versteht man darunter Produkt, die folgenden Merkmale aufweisen:
- relativ einfache, gängige und stabile Konstruktionen mit geringer Weiterentwicklung;
- ihre generische Gerätegruppe ist für ihre Sicherheit bekannt und wurde in der Vergangenheit nicht mit Sicherheitsproblemen in Verbindung gebracht;
- wohlbekannte klinische Leistungsmerkmale und ihre generische Produktgruppe sind Standardprodukte, bei denen sich die Indikationen und der Stand der Technik kaum weiterentwickeln;
- eine lange Historie auf dem Markt.
Daher können alle Produkte, die alle diese Kriterien erfüllen, als "etablierte Technologien" gelten, auch wenn sie nicht in Artikel 61, Absatz 6b der MDR aufgeführt sind.
Das Verfahren der klinische Bewertung
Bei einer klinischen Bewertung müssen systematisch und fortlaufend klinische Daten erhoben, analysiert und bewertet werden, um so die Sicherheit und Leistungsfähigkeit des Medizinproduktes nachweisen zu können.
Laut MDR, Artikel 61, Abschnitt 3 hat eine klinische Bewertung nach einem genau definierten und methodisch fundierten Verfahren abzulaufen, das sich auf folgende Grundlagen stützt:
- eine kritische Bewertung der einschlägigen derzeit verfügbaren wissenschaftlichen Fachliteratur über Sicherheit, Leistung, Auslegungsmerkmale und Zweckbestimmung des Produkts; dabei müssen folgende Bedingungen erfüllt sein:
- das Produkt, das Gegenstand der klinischen Bewertung für die Zweckbestimmung ist, ist dem Produkt, auf das sich die Daten beziehen, gemäß Anhang XIV Abschnitt 3 nachgewiesenermaßen gleichartig und
- die Daten zeigen in geeigneter Weise die Übereinstimmung mit den einschlägigen Sicherheits- und Leistungsanforderungen;
- eine kritische Bewertung der Ergebnisse aller verfügbaren klinischen Prüfungen, wobei gebührend berücksichtigt wird, ob die Prüfungen gemäß den Artikeln 62 bis 80, gemäß nach Artikel 81 erlassenen Rechtsakten und gemäß Anhang XV durchgeführt wurden und
- eine Berücksichtigung der gegebenenfalls derzeit verfügbaren anderen Behandlungsoptionen für diesen Zweck.
Seit Juni 2016 ist das genaue Verfahren in der von der Europäischen Kommission veröffentlichten Guideline zur klinischen Bewertung (MEDDEV 2.7/1 rev. 4) detailliert beschrieben. Neu dabei ist, dass im klinischen Bewertungsplan, der die Ziele und den Aufbau der klinischen Bewertung festlegt, zukünftig auch ein klinischer Entwicklungsplan enthalten sein muss.
Der klinische Entwicklungsplan z. B. beschreibt im Detail die klinische Planung, die vom Hersteller vorgesehen ist, um die klinische Sicherheit, die geringstmögliche Belastung und den effektiven Nutzen des zu bewertenden Medizinprodukts über eigene klinische Daten nachweisen zu können. Die klinische Planung reicht dabei von explorativen über pivotale Studien bis hin zur klinischen Nachbeobachtung nach dem Inverkehrbringen (PMS), unter Angabe von Etappenzielen und der Beschreibung möglicher Akzeptanzkriterien.
Generell gilt laut MDR, dass eine klinische Bewertung und die dazugehörigen Unterlagen während des gesamten Lebenszyklus des Produkts anhand der klinischen Daten zu aktualisieren sind. Dazu benötigt der Hersteller einen Plan für die klinische Nachbeobachtung nach dem Inverkehrbringen gemäß Anhang XIV Teil B der MDR und gemäß Artikel 84 (siehe PMCF-Studien).
Kurz beschrieben, läuft eine klinische Bewertung folgendermaßen ab:
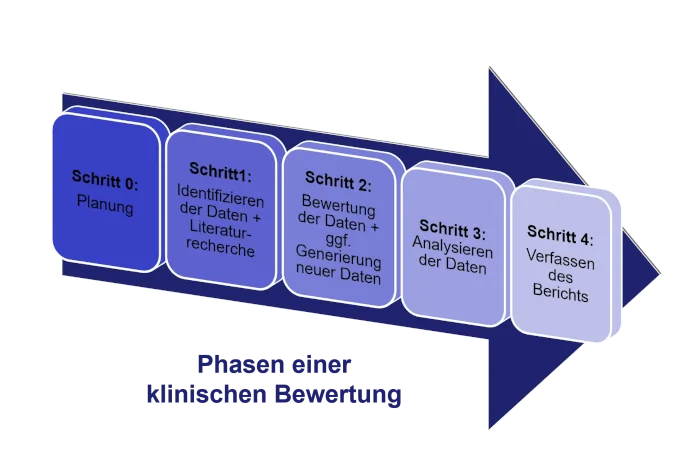
- 0) Planung:
- Festlegen der regulatorischen Anforderungen, welche durch die klinischen Daten zu belegen sind
- 1) Identifizierung:
- Recherche bzgl. aller relevanten Daten aus der wissenschaftlichen Literatur
- Daten vom Hersteller und Daten aus Studien
- State of the Art innerhalb der vorgegebenen Zweckbestimmung des Produkts – was ist im Moment das Standardprodukt für den ausgewählten Zweck – Vergleich mit dem neuen Produkt
- 2) Beurteilung der Daten:
- Auswerten der Einzeldaten
- Beurteilung, ob die verfügbaren Daten für den Nachweis der Produktsicherheit ausreichend sind
- Ggf. weiterer Generierung von klinischen Daten, um offene Punkte in Hinblick auf Sicherheit und Leitung zu klären
- 3) Analyse:
- Erstellen einer zusammenfassenden klinischen Bewertung des Produkts über die Sicherheit und die Leistung
- 4) Bericht
- Abschlussbericht über die Bewertung
Klinische Bewertung - Äquivalenzroute
Das Konzept der Gleichartigkeit mit anderen Produkten kann laut MDR für Produkte, für die bereits klinische Daten vorliegen, auch weiterhin angewendet werden, allerdings nur in einer begrenzten Anzahl von Situationen. Die neuen Regeln sind zudem strenger (MDR, Artikel 61 Absatz 3 und MDR, Anhang XIV, Absatz 3). Sie besagen, dass:
Eine klinische Bewertung sich nur dann auf klinische Daten zu einem Produkt stützen kann, wenn die Gleichartigkeit zwischen dem äquivalenten Produkt und dem eigenem Produkt nachgewiesen werden kann. Zum Nachweis der Gleichartigkeit werden die folgenden technischen, biologischen und klinischen Merkmale herangezogen:
- Technisch: Das Produkt ist von ähnlicher Bauart, wird unter ähnlichen Anwendungsbedingungen angewandt, haben ähnliche Spezifikationen und Eigenschaften einschließlich physikalisch-chemischer Eigenschaften wie Energieintensität, Zugfestigkeit, Viskosität, Oberflächenbeschaffenheit, Wellenlänge und Software-Algorithmen, verwendet gegebenenfalls ähnliche Entwicklungsmethoden und hat ähnliche Funktionsgrundsätze und entscheidende Leistungsanforderungen.
- Biologisch: Das Produkt verwendet die gleichen Materialien oder Stoffe im Kontakt mit den gleichen menschlichen Geweben oder Körperflüssigkeiten für eine ähnliche Art und Dauer des Kontakts bei ähnlichem Abgabeverhalten der Stoffe einschließlich Abbauprodukte und herauslösbarer Bestandteile („leachables“).
- Klinisch: Das Produkt wird unter der gleichen klinischen Bedingung oder zum gleichen klinischen Zweck, einschließlich eines ähnlichen Schweregrads und Stadiums der Krankheit, an der gleichen Körperstelle und bei ähnlichen Patientenpopulationen in Bezug auf u.a. Alter, Anatomie und Physiologie angewandt, hat die gleichen Anwender und erbringt eine ähnliche, maßgebliche und entscheidende Leistung im Hinblick auf die erwartete klinische Wirkung für eine spezielle Zweckbestimmung.
Die im ersten Absatz aufgeführten Merkmale müssen in einer Weise gleichartig sein, dass es keinen klinisch bedeutsamen Unterschied bei der Sicherheit und klinischen Leistung der Produkte gibt. Die Prüfung der Gleichartigkeit stützt sich auf eine angemessene wissenschaftliche Begründung.
Der Nachweis der Äquivalenz kann aber nur gelingen, wenn der Hersteller über entsprechende Daten zu den Äquivalenzprodukten verfügt und dies auch nachweisen kann.
Die MDR besagt dazu in Anhang XIV, Teil A, Abschnitt 3: Es muss eindeutig nachgewiesen werden, dass die Hersteller über einen hinreichenden Zugang zu den Daten von Produkten, mit denen sie die Gleichartigkeit geltend machen, verfügen, um die von ihnen behauptete Gleichartigkeit belegen zu können.
In der MEDDEV 2.7/1 rev. 4 führt in Anhang A1 Voraussetzungen auf, unter welchen die klinische, technische und biologische Äquivalenz als gegeben angenommen werden darf. Der Hersteller wird auch dazu verpflichtet, die Unterschiede zu den Vergleichsprodukten aufzuzeigen und zu erklären, warum diese die klinische Leistungsfähigkeit und die klinische Sicherheit nicht signifikant beeinflussen.
Die MDCG 2020-5 „ Clinical Evaluation – Equivalence” kann als Leitfaden von Herstellern und benannten Stellen genutzt werden, um die Äquivalenz normen-konform durchzuführen. Das Dokument zeigt die Unterscheide zwischen der MEDDEV und der MDR auf, da die Anforderungen der MDR und der MEDDEV an die Äquivalenz nicht deckungsgleich sind.
Um den Nachweis der Gleichwertigkeit zu erbringen, eignet sich am besten eine Tabelle. Dies schlägt auch die MDCG 202-5 vor und stellt Tabellenbeispiele zur Verfügung.
In Fällen, in denen die Gleichwertigkeit gemäß der MDR nicht oder nicht vollständig nachgewiesen werden kann, können die Daten von den Vergleichsprodukten (dann als similar devices) laut MDCG 2020-5 für eine Vielzahl anderer Zwecke nützlich sein, so z. B.:
- für die Ermittlung relevanter Gefahren und klinischer Risiken, um diese in das Risikomanagement aufzunehmen.
- zum Verstehen des Stands der Technik / Wissenschaft, des natürlichen Krankheitsverlaufs und der alternativen Behandlungsmöglichkeiten.
- zur Unterstützung bei der Festlegung des Umfangs der klinischen Bewertung durch Ermittlung von Konstruktionsmerkmalen ähnlicher Produkte, die besondere Leistungs- oder Sicherheitsbedenken aufwerfen.
- als Beiträge zur Gestaltung von klinischen Prüfungen, dem PMS-System und dem PMCF-Prozess.
- für die Identifizierung relevanter und spezifizierter klinischer Ergebnisparameter für den angestrebten klinischen Nutzen auf der Grundlage der veröffentlichten klinischen Daten zu dem/ den ähnlichen Produkt(en).
- für die Festlegung von Mindestanforderungen für einen quantifizierten klinischen Nutzen, der als klinisch relevant angesehen wird, und/oder zur Ermittlung akzeptabler Inzidenzraten von Risiken und unerwünschten Ereignissen.
Bitte beachten Sie, dass die MDCG-Leitfäden keinen bindenden Charakter für Behörden oder benannte Stellen haben. Die momentane Praxis zeigt jedoch, dass den Vorschlägen dieser Dokumente in den allermeisten Fällen gefolgt wird.
Zusammengefasst
Da die Bewertung nach dem Äquivalenzprinzip mit der MDR stark eingeschränkt wird, bedeutet das, dass Sie als Hersteller mehr eigene klinische Daten für Ihre Produkte generieren und aufrechterhalten müssen, um im Rahmen der klinischen Bewertung klinische Evidenz mit den grundlegenden Sicherheits- und Leistungsanforderungen zeigen zu können. Dies müssen Sie im klinischen Entwicklungsplan entsprechend berücksichtigen.
Zudem werden aufgrund der Einschränkungen der Äquivalenzbetrachtung und den hohen Anforderungen an die Qualität der klinischen Daten klinische Prüfungen für das erstmalige Inverkehrbringen eines Medizinprodukts und PMCF-Studien für die Re-Zertifizierung immer mehr erforderlich sein wird. Für Klasse III und implantierbare Produkte sind diese bis auf wenige Ausnahmen sogar verpflichtend durchzuführen.
Daher sollten Sie als Hersteller, Ihre eigenen klinischen Daten für die Produkte dahingehend kritisch überprüfen. Sollten Sie Lücken finden, feststellen, dass Ihre Produkte unter der MDR einer anderen Klasse zugeordnet werden oder Sie Ihre Produkte zum ersten Mal klinisch bewerten müssen, sollten Sie aktiv klinische Daten zu den eigenen Produkten über klinische Prüfungen oder PMCF-Studien generieren.
Für etwas Entspannung bei den Herstellern von Bestandsmedizinprodukten sorgen die Veröffentlichungen der EU Koordinierungsgruppe Medizinprodukte (MDCG). Insbesondere MDCG 2020-6 und MDCG 2021-25 konkretisieren die anwendbaren Quellen für klinische Daten bei „Altgeräten“ (legacy devices). Die Anforderungen der MDR an das PMS und wenn nötig an das PMCF müssen trotzdem voll umfänglich erfüllt werden.
Klinische Bewertung von Bestandsprodukten (Legacy devices)
Für Bestandsprodukte, die sogenannten Legacy devices, hat die MDCG das Dokument MDCG 2020-6: „Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC“ veröffentlicht. Es dient als Leitfaden für klinische Daten, die ausreichen, um die Konformität der Altprodukte (Legacy devices) mit den einschlägigen allgemeinen Sicherheits- und Leistungsanforderungen (GSPR) gemäß Artikel 61 Absatz 1 der MDR nachzuweisen.
Auch hier ist zu beachten, dass diese Leitfäden keinen bindenden Charakter für Behörden oder benannte Stellen haben.
Unter Legacy devices versteht man laut MDCG 2021-25 Produkte, die gemäß Artikel 120 Absatz 3 der MDR nach dem Anwendungsdatum der MDR und bis zum 26. Mai 2024 in Verkehr gebracht werden, wenn bestimmte Bedingungen erfüllt sind. Es kann sich dabei um folgende Produkte handeln:
- Produkte der Klasse I gemäß der Richtlinie 93/42/EWG (MDD), für die vor dem 26. Mai 2021 eine EG-Konformitätserklärung ausgestellt wurde und für die das Konformitätsbewertungsverfahren gemäß der MDR die Einschaltung einer benannten Stelle erfordert;
- Produkte, für die eine gültige EG-Bescheinigung vorliegt, die gemäß der Richtlinie 90/385/EWG (AIMDD) oder der MDD vor dem 26. Mai 2021 ausgestellt wurde.
Als "alte" Produkte (Old devices) gelten Produkte, die vor dem 26. Mai 2021 gemäß der AIMDD oder der MDD oder gemäß den vor dem Inkrafttreten der Richtlinien geltenden Vorschriften in Verkehr gebracht wurden.
Der Begriff „etablierte Technologie“ (well-established technology) wird in Artikel 52 Absatz 5 und Artikel 61 Absatz 8 der MDR verwendet, ist aber in diesen Artikeln nicht definiert. Der Begriff ist nicht auf die in Artikel 61 Absatz 6b aufgeführten Produkte beschränkt; in Artikel 61 Absatz 8 heißt es ausdrücklich, dass dies auch Produkte einschließt, die den in Artikel 61 Absatz 6b aufgeführten freigestellten Produkten ähneln und die in Zukunft in diese Liste aufgenommen werden könnten. Die gemeinsamen Merkmale der Produkte, bei denen es sich um etablierte Technologien handelt, sind folgende:
- relativ einfache, gängige und stabile Konstruktionen mit geringer Weiterentwicklung
- ihre generische Gerätegruppe ist für ihre Sicherheit bekannt und wurde in der Vergangenheit nicht mit Sicherheitsproblemen in Verbindung gebracht;
- bekannte klinische Leistungsmerkmale und ihre generische Produktgruppe sind Standardprodukte, bei denen es nur wenig Entwicklung bei Indikationen und dem Stand der Technik gibt;
- eine lange Geschichte auf dem Markt.
Daher können alle Produkte, die alle diese Kriterien erfüllen, als "etablierte Technologien" gelten (MDCG 2020-6).
Laut MDCG 2020-6 ist bei der Bewertung der Konformität von Altprodukten im Rahmen der MDR zu prüfen, ob PMCF-Studien, die im Rahmen der MDD/AIMDD (und gegebenenfalls während des Übergangszeitraums im Rahmen der MDR) als notwendig erachtet wurden, ordnungsgemäß durchgeführt und die Ergebnisse bei der klinischen Bewertung im Rahmen der MDR in vollem Umfang berücksichtigt wurden.
Die Hersteller sind verpflichtet, einen Plan für die klinische Bewertung zu dokumentieren, um die Anforderungen von MDR Anhang XIV Abschnitt 1a zu erfüllen.
Laut MDCG 2020-6 sollte ein modifizierter klinischer Bewertungsplan für Altprodukte mindestens folgendes enthalten:
- Eine Identifizierung der GSPR, die durch relevante klinische Daten unterstützt werden müssen.
- Eine Spezifikation der Zweckbestimmung des Produkts.
- Eine klare Spezifikation der vorgesehenen Zielgruppen mit eindeutigen Indikationen und Kontraindikationen.
- Eine detaillierte Beschreibung des beabsichtigten klinischen Nutzens für die Patienten mit relevanten und spezifizierten klinischen Ergebnisparametern.
- Eine Strategie zur Ermittlung, Analyse und Bewertung alternativer Behandlungen.
- Eine Spezifikation der Methoden, die für die Untersuchung der qualitativen und quantitativen Aspekte der klinischen Sicherheit mit eindeutigem Bezug auf die Bestimmung von Restrisiken und Nebenwirkungen zeigt.
- Eine indikative Liste und Spezifikation von Parametern, anhand derer auf der Grundlage des Stands der Technik / Wissenschaft die Annehmbarkeit des Nutzen-Risiko-Verhältnisses für die verschiedenen Indikationen und für die Zweckbestimmung(en) des Produkts bestimmt werden kann.
- Angaben darüber, wie Nutzen-Risiko-Probleme im Zusammenhang mit bestimmten Bestandteilen, z. B. der Verwendung von pharmazeutischem, nicht lebensfähigem tierischem oder menschlichem Gewebe, behandelt werden sollen.
- Eine Strategie und Methodik zur Ermittlung, Analyse und Bewertung aller relevanten verfügbaren klinischen Daten unter Berücksichtigung der geänderten Definition für klinische Daten.
- Nachweis der Gleichwertigkeit, wenn die klinischen Daten eines gleichwertigen Produkts in die klinische Bewertung einbezogen werden.
- Eine Definition des erforderlichen Niveaus des klinischen Nachweises, dass angesichts der Merkmale des Produkts und seiner Zweckbestimmung angemessen sein muss.
- Eine Strategie und Methodik zur systematischen Sammlung, Zusammenfassung und Bewertung von Daten aus der Überwachung nach dem Inverkehrbringen, um die kontinuierliche Sicherheit und Leistung nachzuweisen, und
- inwieweit bei den Altprodukten Beanstandungen in Bezug auf Sicherheit und Leistung aufgetreten sind.
Zu den Quellen für klinische Daten vor dem Inverkehrbringen von Altprodukten können gehören (MDCG 2020-6):
- Klinische Prüfberichte über das betreffende Produkt;
- Berichte über klinische Prüfungen oder andere in der wissenschaftlichen Literatur veröffentlichte Studien zu einem Produkt, für das die Gleichwertigkeit mit dem betreffenden Produkt gemäß der MDR nachgewiesen werden kann;
- In der wissenschaftlichen Fachliteratur veröffentlichte Berichte über sonstige klinische Erfahrungen mit dem betreffenden Produkt oder einem Produkt, für das die Gleichwertigkeit mit dem betreffenden Produkt nachgewiesen werden kann;
- Sonstige Daten aus der Zeit vor dem Inverkehrbringen, z. B. Fallberichte über Erfahrungen mit der Verwendung des betreffenden Produkts, wie z. B. Berichte über die Anwendung im Rahmen von Mitleidsmaßnahmen oder humanitären Ausnahmefällen. Beachten Sie, dass diese Art von Daten vor dem Inverkehrbringen anfälliger für Verzerrungen sein können, als die oben genannten.
Quellen für klinische Daten nach dem Inverkehrbringen beziehen sich auf Daten, die nach der ersten CE-Kennzeichnung gemäß den Richtlinien (oder vor der Einführung einer neuen Indikation oder Auslegungsvariante) erhoben wurden. Diese können umfassen:
- PMS-Daten, Berichte über Beschwerden und Zwischenfälle;
- PMCF-Studien, einschließlich klinischer Prüfungen nach dem Inverkehrbringen;
- Unabhängige klinische Studien, die mit dem Produkt durchgeführt wurden;
- Geräteregister;
- Aus der Literatur entnommene Daten.
Bei gut eingeführten Technologien (well-established technology) kann die klinische Bewertung auf Daten beruhen, die von ähnlichen Produkten stammen (siehe Abschnitt 6.5 (e) der MDCG 2020-6) genannten Bedingungen. Wenn klinische Daten von gleichwertigen Produkten verwendet werden, muss die Gleichwertigkeit gemäß den Anforderungen der MDR nachgewiesen werden. Die MDCG 2020-6 schlägt auch eine Hierarchie der klinischen Nachweise zur Bestätigung der Konformität mit den einschlägigen GSPR gemäß der MDR vor, siehe hierzu Appendix II des Leitfadens.
Klasse III und Implantierbare Produkte – Bewertung und klinische Prüfungen
Laut MDR Artikel 61, Absatz 4 müssen für implantierbare Produkte und Produkte der Klasse III klinische Prüfungen durchgeführt werden, es sei denn:
- das betreffende Produkt wurde durch Änderungen eines bereits von demselben Hersteller in Verkehr gebrachten Produkts konzipiert,
- der Hersteller hat nachgewiesen, dass das geänderte Produkt dem in Verkehr gebrachten Produkt gemäß Anhang XIV Abschnitt 3 gleichartig ist, und dieser Nachweis ist von der benannten Stelle bestätigt worden und
- die klinische Bewertung des in Verkehr gebrachten Produkts reicht aus, um nachzuweisen, dass das geänderte Produkt die einschlägigen Sicherheits- und Leistungsanforderungen erfüllt.
In diesem Fall prüft die benannte Stelle, dass der Plan für die klinische Nachbeobachtung nach dem Inverkehrbringen zweckdienlich ist und Studien nach dem Inverkehrbringen beinhaltet, um Sicherheit und Leistung des Produkts nachzuweisen (MDR, Artikel 61, Absatz 4).
Artikel 61, Absatz 5 und 6 besagen, dass ein Hersteller eines Produkts, das nachweislich einem bereits in Verkehr gebrachten, nicht von ihm hergestellten Produkts gleichartig ist, sich ebenfalls auf Absatz 4 berufen kann, um keine klinische Prüfung durchführen zu müssen, sofern zusätzlich zu den Anforderungen des genannten Absatzes die folgenden Bedingungen erfüllt sind:
- Die beiden Hersteller haben einen Vertrag geschlossen, in dem dem Hersteller des zweiten Produkts ausdrücklich der uneingeschränkte Zugang zur technischen Dokumentation durchgängig gestattet wird, und
- die ursprüngliche klinische Bewertung wurde unter Einhaltung der Anforderungen der vorliegenden Verordnung durchgeführt
- und der Hersteller des zweiten Produkts liefert der benannten Stelle den eindeutigen Nachweis hierfür.
Es gilt weiter: Die Anforderung, klinische Prüfungen gemäß Absatz 4 durchzuführen, gilt nicht für implantierbare Produkte und Produkte der Klasse III,
a) die gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG rechtmäßig in Verkehr gebracht oder in Betrieb genommen wurden und deren klinische Bewertung
- sich auf ausreichende klinische Daten stützt und
- mit den einschlägigen produktspezifischen Spezifikationen für die klinische Bewertung dieser Art von Produkten im Einklang steht, sofern diese gemeinsamen Spezifikation verfügbar sind, oder
b) bei denen es sich um Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Zahn- bzw. Knochenplatten, Drähte, Stifte, Klemmen oder Verbindungsstücke handelt, deren klinische Bewertung auf der Grundlage ausreichender klinischer Daten erfolgt und mit den einschlägigen produktspezifischen Spezifikationen im Einklang steht, sofern diese Spezifikationen verfügbar sind.
Generell gilt, dass man nur noch unter ganz bestimmten Voraussetzungen auf eine klinische Bewertung verzichten kann. Artikel 61, Absatz 10 der MDR sagt dazu:
Wird der Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungs-anforderungen auf der Grundlage klinischer Daten für ungeeignet erachtet, ist jede solche Ausnahme auf der Grundlage des Risikomanagements des Herstellers und unter Berücksichtigung der besonderen Merkmale des Zusammenspiels zwischen dem Produkt und dem menschlichen Körper, der bezweckten klinischen Leistung und der Angaben des Herstellers angemessen zu begründen; dies gilt unbeschadet des Absatzes 4. In diesem Fall muss der Hersteller in der technischen Dokumentation gemäß Anhang II gebührend begründen, warum er den Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen allein auf der Grundlage der Ergebnisse nicht klinischer Testmethoden, einschließlich Leistungsbewertung, technischer Prüfung („bench testing“) und präklinischer Bewertung, für geeignet hält.
Sie als Hersteller dürfen im Prinzip nur noch bei ganz unkritischen Medizinprodukten auf eine klinische Bewertung verzichten.
Klinische Bewertung und geänderte Übergangsfristen
Am 15.03.2023, rechtskräftig mit Veröffentlichung im Amtsblatt am 20.03.2023, hat das europäische Parlament final beschlossen (3. Corringendum), dass die in Artikel 120 der MDR geregelten Übergangsfristen erneut verlängert werden. Die Änderungen werden in der Verordnung (EU) 2023/607 geregelt (siehe: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32023R0607).Hersteller von sogenannten Bestandsprodukten (Legacy devices) können ihre Produkte daher bis zum Ende der neuen Übergangsfristen weiter Inverkehr bringen, ohne einen Konformitätsnachweis nach EU-Verordnung 2017/745 (MDR) zu erbringen. Dies betrifft auch die klinische Bewertung.
Die neuen Übergangsfristen sind für Produkte der:
- Klasse III und für implantierbare Produkte der Klasse IIb - 31.12.2027
- Andere Produkte der Klassen IIb, IIa und Is und Im – 31.12.2028
- Klasse Ir – 31.12.2028.
Um als Hersteller diese neuen Übergangszeiten nutzen zu können, müssen jedoch einige Bedingungen erfüllt werden (siehe https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32023R0607). Andere Anforderungen wie z. B. die Überwachung nach dem Inverkehrbringen, die Vigilanz und das Aufbringen der UDI sind von dieser Verlängerung nicht betroffen.
Bedingungen für die Verlängerung
Nach Verordnung (EU) 2023/607 können die oben genannten Fristverlängerungen der MDR jedoch nur in Anspruch genommen werden, wenn bei den bestehenden Zertifikaten folgende Voraussetzungen erfüllt sind:
- die MDD-Zertifikate waren am 26.05.2021 noch gültig und wurden im Anschluss nicht zurückgezogen – die Zertifikate behalten Ihre Gültigkeit bis zum Ablauf der Übergangsfrist
- MDD-Zertifikate waren am 26.05.2021 noch gültig und sind vor dem 20.3.2023 abgelaufen – die Zertifikate behalten Ihre Gültigkeit, wenn:
- eine schriftliche Vereinbarung zwischen dem Medizinproduktehersteller und der Benannten Stelle vor dem Ablaufdatum des Zertifikats unterzeichnet wurde. Die Vereinbarung betrifft die Konformitätsbewertung des Medizinprodukts bzw. des Produkts, das das ursprüngliche Medizinprodukt ersetzen soll, oder
- ine Benannte Stelle eines Mitgliedstaats eine Ausnahme von dem in der MDR vorgesehenen Konformitätsbewertungsverfahrens nach Art. 59 Abs. 1 MDR oder eine Ausnahme gegenüber dem Hersteller nach Art. 97 Abs. 1 MDR, ein solches durchzuführen, gewährt hat.
Wenn die Voraussetzungen bei den Zertifikaten erfüllt sind, darf die Fristverlängerung für die Produkte nur in Anspruch genommen werden, wenn (Verordnung (EU) 2023/607):
- die Produkte weiterhin die Anforderungen der Richtlinie 93/42/EWG (MDD) erfüllen
- keine wesentlichen Änderungen bei der Auslegung und der Zweckbestimmung vorgenommen wurden
- die Produkte kein unannehmbares Risiko für die Gesundheit oder Sicherheit der Patienten, Anwender oder anderer Personen oder für andere Aspekte des Schutzes der öffentlichen Gesundheit darstellen
- der Hersteller spätestens am 26. Mai 2024 ein Qualitätsmanagementsystem eingerichtet hat, und
- der Hersteller bis spätestens 26. Mai 2024 einen förmlichen Antrag zur Konformitätsbewertung bei einer benannten Stelle eingereicht hat und bis spätestens 26. September 2024 eine schriftliche Vereinbarung mit der Benannten Stelle unterzeichnet hat.
Quellen:
- NAKI - Nationaler Arbeitskreis des Bundesgesundheitsministeriums ( Link )
- Volltext der konsolidierten Fassung der MDR (nicht amtlich)
- Volltext der Grundfassung
- MDCG 2020-5 Clinical Evaluation - Equivalence A guide for manufacturers and notified bodies
- MDCG 2020-6 Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC A guide for manufacturers and notified bodies
- MDCG 2021-25 Regulation (EU) 2017/745 - application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC
Klinische Bewertung (CER)
Regulatorisches zur klinischen Bewertung FAQ
Bei der klinischen Bewertung handelt es sich um einen systematischen und fortlaufenden Prozess, der sowohl in der Phase der Produktentwicklung, als auch nach dem Markteintritt des Produktes bis hin zum Ende des Produktlebenszyklus eine Rolle spielt.
Im Rahmen der klinischen Bewertung können wir Sie daher mit folgenden Dienstleistungen unterstützen, so dass Ihr Produkt schnell die Zulassung bekommt und Ihre Dokumentation auch nach dem Markteintritt immer aktuell ist:
- Überprüfung, ob vor der Zulassung eine klinische Prüfung notwendig ist und wenn ja, in welchem Umfang diese erfolgen sollte.
- Erstellen des klinischen Bewertungsplans inkl. klinischem Entwicklungsplan für Ihre strategische Planung.
- Überprüfung, Neuerstellung oder Überarbeitung / Aktualisierung Ihrer klinischen Bewertung.
- Regelmäßige Überprüfung des Stands der Technik (State of the Art), so dass Sie immer auf potentielle Weiter- und Neuentwicklungen reagieren können
- Durchführen der Literaturrecherche anhand einer nachvollziehbaren Suchstrategie (wie z. B. der PICO-Suche), inkl. Zusammenfassung und Bewertung. Beurteilung der klinischen Daten anhand von Bewertungsschemen, wie z. B. JADAD, MINORS, usw.
- Updateservice für Ihre klinische Bewertung.
- Überprüfung in regelmäßigen Abständen auf neue Literatur.
- Überprüfung des Stands der Technik (SotA), so dass Sie schnell auf Neu- und Weiterentwicklungen reagieren können.
- Überprüfungen der Datenbanken für Vorkommnisse (BfArM, FDA (MAUDE und TPLC) und weitere).
- Beratung zu nötigen Updates.
Mit unserer Hilfe können Sie:
- Unnötige Arbeiten vermeiden
- Ihre Risikoanalyse regelmäßig an neu auftretende Risiken anpassen
- Zeit und Kosten sparen, indem Sie die aufwendige Recherchearbeit an einer Stelle bündeln
Haben wir Ihr Interesse geweckt, dann kontaktieren Sie uns. Rufen Sie uns an, schreiben Sie uns oder füllen Sie unser Kontaktformular aus. Wir freuen uns auf Sie.
- Wir arbeiten normenkonform nach MDR, MedDev 2.7/1 Rev. 4 und der MDD
- Wir fertigen Ihre Dokumente schnell und zuverlässig in Deutsch oder Englisch an und nutzen dazu entweder Ihre oder die AnCura-eigenen normenkonformen Formblätter
- Wir erfüllen die Anforderungen an die Autoren klinischer Bewertungen
- Wir arbeiten mit einem Netzwerk von Medizinern / Klinikern / Statistikern zusammen.
- Gemeinsam finden wir eine Lösung. Ihre Zufriedenheit ist unser oberstes Ziel!
Regulatorisches zur klinischen Bewertung
Unsere Leistungen - Klinische Bewertung (KB)
Wir unterstützen Sie als Hersteller in jeder Phase der klinischen Bewertung Ihres Medizinproduktes vom Aufstellen des klinischen Bewertungsplans, über das Durchführen der klinischen Bewertung bis hin zur geplanten und/oder auf Vorkommnisse basierenden Aktualisierung Ihrer Bewertung. Wir erstellen klinische Bewertungen für Medizinprodukte der Klassen I, II a, IIb, b, III an.
Dies beinhaltet folgende Leistungen:
Erstellen des klinischen Bewertungsplans (CEP), Planung der klinischen Bewertung
Wir helfen oder erstellen Ihnen den, von der MDR geforderten, klinischen Bewertungsplan (CEP). In diesem müssen die genauen Ziele und der Aufbau der klinischen Bewertung beschrieben werden.Erstellen des klinischen Entwicklungsplans
Der CEP muss auch einen klinischen Entwicklungsplan enthalten. Dieser beschreibt, wie Sie als Hersteller die klinische Sicherheit, die geringstmögliche Belastung und den effektiven Nutzen Ihres Medizinproduktes anhand von eigenen klinischen Daten nachweisen wollen. Er beinhaltet die geplanten Etappenziele und die Kriterien für die mögliche Akzeptanz. Möglich sind explorative Studien, Pilotstudien bis hin zur klinischen Überwachung nach dem Inverkehrbringen (PMS). Wir unterstützen Sie, indem wir in enger Absprache mit Ihnen, die Pläne (CEP, PMS, PMCF) aufstellen, Etappenziele erarbeiten und mögliche Studien für Sie planen.Literaturrecherche
Laut MDR muss eine klinische Bewertung nach einem genau definierten und methodisch fundierten Verfahren erfolgen, das sich auf folgende Grundlagen stützt: a) eine kritische Bewertung der einschlägigen derzeit verfügbaren wissenschaftlichen Fachliteratur über Sicherheit, Leistung, Auslegungsmerkmale und Zweckbestimmung des Produkts. Auch hier unterstützen wir Sie gerne, da die Suche in den einschlägigen Datenbanken wie z.B. Embase erfolgen muss und viel Zeit in Anspruch nimmt. Dazu gehört das Erstellen des Rechercheplans, die Durchführung der systematischen Suche in den einschlägigen Datenbanken, das Zusammenfassen der Daten und eine Empfehlung, ob diese für eine klinische Bewertung Ihres Produktes ausreichend sind oder ob Sie selber noch Daten generieren sollten.Durchführung der klinischen Bewertung für Ihr Medizinprodukt
Dies umfasst alle Schritte einer klinischen Bewertung von der Planung (Suchstrategie, usw.), über das Identifizieren und Suchen der relevanten klinischen Daten, der Bewertung der vorhandenen Daten (Daten vom Hersteller und aus der Literatur), sowie einer Empfehlung an Sie neue Daten zu generieren falls nötig, bis hin zur Analyse der Daten und dem Verfassen des Abschlussberichts. Dabei ist es uns wichtig, Sie als Hersteller ganz individuell zu unterstützen. Da die Anforderungen an die Autoren einer klinischen Bewertung mit der MDR verschärft wurden und vor allem viele kleine und mittlere Unternehmen nicht über das entsprechende Personal verfügen, können Sie uns mit der Planung und dem Erstellen Ihrer klinischen Bewertung beauftragen. Dies führt zu einer Zeit-, Kosten- und Ressourcenersparnis für Sie als Hersteller und Sie sich ganz auf Ihre Stärken, dem Entwickeln innovativer Medizinprodukte, konzentrieren.Aktualisierung Ihrer klinischen Bewertung
Die Aktualisierung der klinischen Bewertung wird von der MDR vorgeschrieben. Generell gilt laut MDR, dass eine klinische Bewertung und die dazugehörigen Unterlagen während des gesamten Lebenszyklus des Produkts anhand der klinischen Daten zu aktualisieren sind. Auf Wunsch führen wir in regelmäßigen Abständen bzw. bei Bedarf nach z.B. Vorkommnissen eine Literatursuche für Sie durch und machen Sie auf neue klinische Daten aufmerksam. Bei Bedarf können wir dann auch die klinische Bewertung aktualisieren bzw. einen Bericht verfassen, warum eine Aktualisierung der Bewertung nicht notwendig ist.Überprüfen Ihrer klinischen Bewertung
Sollte Ihnen als Kleinst- oder Kleinunternehmen das in der MDR-geforderte Fachpersonal fehlen, Sie möchten aber trotzdem Ihre klinische Bewertung selber erstellen, können wir diese im Anschluss für Sie überprüfen, ob alle Anforderungen der MDR und der MedDev 2.7/1 Rev. 4 erfüllt worden sind. Hier bieten sich eine Vielzahl an Möglichkeiten, kontaktieren Sie uns und wir finden eine Lösung.
Hierfür nutzen wir unsere eigenen, nach der MedDev 2.7/1 Rev. 4 erstellten Formblätter, die wir Ihnen auch gerne zur Verfügung stellen. Erstellt wurden diese nach den Richtlinien der DIN EN ISO 9001, was bedeutet, dass wir Sie regelmäßig überprüfen und aktualisieren.
Generell bieten wir unsere Leistungen sowohl in Deutsch, als auch in Englisch an.
Kontaktieren Sie uns und wir erstellen Ihnen gerne ein angepasstes Angebt.
FAQ
Was ist eine klinische Bewertung von Medizinprodukten?
Die klinische Bewertung ist ein methodisch fundierter, fortlaufender Prozess zur Erhebung, Bewertung und Analyse klinischer Daten zu einem Medizinprodukt. So soll nachgewiesen werden, dass Nutzen, Leistung und Sicherheit der Medizinprodukte in ausreichendem Maße gegeben sind.
Aus welchen Dokumenten besteht eine Klinische Bewertung?
Eine klinische Bewertung setzt sich u.a. aus dem klinischen Bewertungsplan inkl. dem klinischen Entwicklungsplan (CEP) und der klinischen Bewertung (CER) zusammen. Außerdem sollte ein Plan und Bericht zur Literaturrecherche entweder als gesondertes Dokument oder als Teil des CEP und CER vorliegen.
Wer darf eine klinische Bewertung schreiben / erstellen?
Die klinische Bewertung sollte von einer entsprechend qualifizierten Person oder einem Team durchgeführt werden. Die Person oder das Team sollte u.a Erfahrungen im Bereich der Forschungsmethodik und den regulatorischen Anforderungen haben und z. B. Kenntnisse über die Produkttechnologie und das Fachwissen in dem betreffenden medizinischen Fachgebiet haben, und über eine entsprechende Ausbildung (Studium, Berufserfahrung) verfügen (Siehe MDR, Artikel 15 oder MEDDEV 2.71 / rev.4, Abschnitt 6.4)
Gibt es Ausnahmen für die Pflicht zur Erstellung einer klinischen Bewertung?
Laut MDR gibt es keine Ausnahmen. Alle Hersteller müssen eine klinische Bewertung für Ihre Produkte planen (Klinischer Bewertungsplan), durchführen und dokumentieren (klinischer Bewertungsbericht). Prüfen Sie vorab, ob es sich um ein Medizinprodukt handelt. Der Umfang der klinischen Bewertung richtet sich nach den Produktmerkmalen und der Zweckbestimmung. Bei einigen Produkten (z. B. einige legacy devices, well-established technologies) besteht die Möglichkeit, die Bewertung nur über die generellen Sicherheits- und Leistungsanforderungen (GSPR) durchzuführen.